DNA mitochondrialne, mtDNA –
materiał genetyczny
w postaci kolistego
DNA
znajdujący się w
macierzy mitochondrium
(
łac.
matrix). Obecność DNA tłumaczona jest teorią
endosymbiotycznego
pochodzenia tych
organelli
.
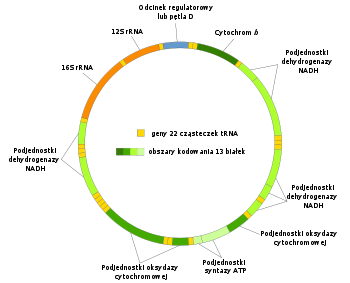
Pojedyncze ludzkie
mitochondrium
zawiera 4–10 kolistych cząsteczek DNA o długości 16569
par zasad
, z których każda koduje 37
genów
. 13 z nich to geny kodujące
białka
, 22 kodują
transferowe RNA
(tRNA), a dwa ostatnie –
rybosomalne RNA
(rRNA). Białka kodowane przez mtDNA to część mitochondrialnych białek
łańcucha oddechowego
, jednak większość białek wchodzących w jego skład jest kodowana przez
genom jądrowy
.
Kod genetyczny
mitochondriów różni się od kodu genetycznego w genomie jądrowym. UGA (jądrowy
kodon
STOP) w mitochondriach oznacza
tryptofan
, AUA (
izoleucyna
) –
metioninę
, a AGA i AGG (
arginina
) są mitochondrialnymi kodonami STOP. Geny mitochondrialne ludzi nie zawierają
intronów
. Nici pojedynczej cząsteczki mtDNA są oznaczane jako H (
ang.
heavy – ciężka) i L (ang. light – lekka). Geny leżą na obydwu niciach. Na nici lekkiej
transkrypcja
rozpoczyna się z jednego
promotora
, a na nici ciężkiej z dwóch.
Dziedziczenie genomu mitochondrialnego
Różne mitochondria w tej samej komórce mogą zawierać różniące się od siebie cząsteczki mtDNA, nawet w pojedynczym mitochondrium nie wszystkie cząsteczki muszą być jednakowe. To rzadkie zjawisko nazywa się
heteroplazmią
. U ssaków mitochondria wraz ze znajdującym się w nich mtDNA są przekazywane następnym pokoleniom niemal wyłącznie w linii żeńskiej. U myszy zaobserwowano, że pochodzące od ojca mitochondria (wraz z zawartym w nich DNA) są niszczone we wczesnych fazach rozwoju
zygoty
. U innych organizmów wygląda to inaczej, np. u ogórka mitochondria dziedziczą się w linii męskiej. Podczas podziału komórki mitochondria rozdzielane są losowo do potomnych komórek.
Choroby mitochondrialne
Mutacje
w genach mitochondrialnych powodują
choroby genetyczne
, których objawy dotyczą głównie tkanek o największym zapotrzebowaniu energetycznym - mięśniowej i nerwowej. Choroby te mają charakterystyczny, matczyny wzór dziedziczenia. Ponadto zróżnicowanie mtDNA w poszczególnych mitochondriach i komórkach wpływa na nasilenie objawów choroby. Przykładami chorób związanych z mutacjami w genomie mitochondrialnym są: LHON – dziedziczna neuropatia wzrokowa Lebera, NARP – neuropatia obwodowa z ataksją i barwnikowym zwyrodnieniem siatkówki, MERF – padaczka miokloniczna z nieprawidłowymi czerwonymi włóknami mięśniowymi,
zespół MELAS
– encefalopatia mitochondrialna z kwasicą mleczanową i epizodami podobnymi do udaru mózgu,
zespół Leigha
. Szacuje się, że na choroby mitochondrialne zapada 1 na 15 000 osób. Leczenie chorób mitochondrialnych jest objawowe.
Zastosowania genomu mitochondrialnego w nauce
DNA mitochondrialny jest wykorzystywany w
genetyce populacyjnej
[1] i medycynie sądowej. Służą do tego obszary hiperzmienne mitochondrialnego DNA – niekodujące fragmenty genomu mitochondrialnego o wysokim zróżnicowaniu u poszczególnych ludzi. Hiperzmienny region 1 (HVR1) obejmuje sekwencję 16024-16365, a hiperzmienny region 2 – sekwencję 73-340. Porównanie DNA mitochondriów pochodzących od ludzi wywodzących się z różnych grup etnicznych pozwoliło na obliczenie, kiedy żyła
mitochondrialna Ewa
– kobieta, od której wywodzą się wszyscy współcześni ludzie (a właściwie ich genom mitochondrialny). Ze względu na obfitość mitochondrialnego
aDNA
w komórkach częściej udaje się uzyskać go z materiałów kopalnych. Dzięki temu poznano genom mitochondrialny
neandertalczyka
wcześniej, niż zaczęto sekwencjonować jego genom jądrowy. W genomie mitochondrialnym neandertalczyka nie znaleziono sekwencji hiperprzmiennych występujących współcześnie.
Zobacz też
Przypisy
Bibliografia
Linki zewnętrzne