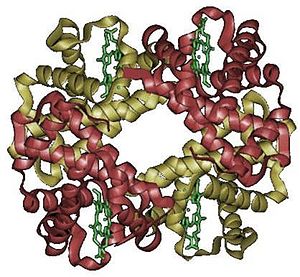
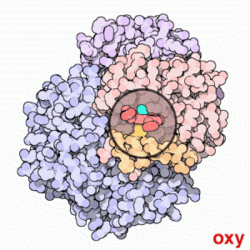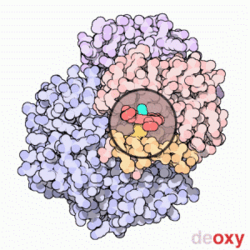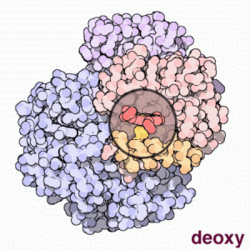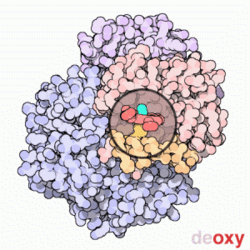
Model cząsteczki hemoglobiny. Cztery zasocjowane podjednostki, z których każda zawiera cząsteczkę hemu (zaznaczoną na zielono). Nie pokazano grup bocznych aminokwasów, a tylko konformację łańcuchów peptydowych – tzw. model wstęgowy
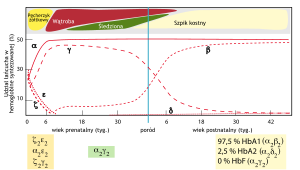
Miejsce i odsetek syntezy łańcuchów hemoglobiny w życiu pre- i postnatalnym
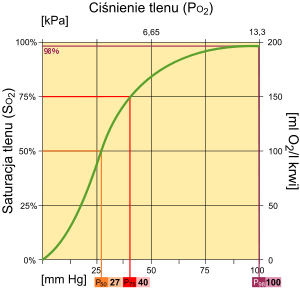
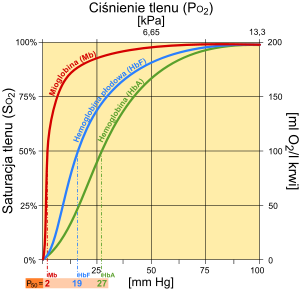
Krzywa wysycenia hemoglobiny w zależności od ciśnienia cząstkowego tlenu.
Porównanie krzywych saturacji Mioglobiny(Mb) z Hemoglobiną (HbA), Hemoglobiną płodową
Hemoglobina, oznaczana też skrótami Hb lub HGB – czerwony barwnik
krwi
,
białko
zawarte w
erytrocytach
, którego zasadniczą funkcją jest przenoszenie
tlenu
– przyłączanie go w płucach i uwalnianie w tkankach.
Mutacje
genu
hemoglobiny prowadzą do chorób dziedzicznych:
anemii sierpowatej
,
talasemii
lub rzadkich chorób zwanych
hemoglobinopatiami
.
Budowa hemoglobiny
Cząsteczka hemoglobiny jest
tetramerem
złożonym z dwóch par białkowych podjednostek. Podjednostki oznaczone są najczęściej literami
greckiego alfabetu
(np. α,β,γ,δ).
| Nazwa hemoglobiny | Pary podjednostek |
|---|
| HbA | α2β2 |
| HbF | α2γ2 |
| HbA2 | α2δ2 |
|
HbS
| α2S2 |
Podjednostki nie są związane
kowalencyjnie
. Każda podjednostka zawiera jako
grupę prostetyczną
(niebiałkową) cząsteczkę
hemu
. Cząsteczka hemu zawiera położony centralnie atom żelaza (Fe2+) umożliwiający jej wiązanie cząsteczek tlenu (O2). Jedna cząsteczka hemoglobiny może przyłączyć od jednej do czterech cząsteczek tlenu, co powoduje że hemoglobina może występować albo w stanie "odtlenowanym" (deoxyHb) lub w różnym stopniu "utlenowania" (oxyHb). Hem nadaje białku (i krwi) czerwony kolor.
Konformacja łańcuchów α i β ludzkiej hemoglobiny wykazuje podobieństwa do cząsteczki
mioglobiny
.
Podział hemoglobin
Hemoglobiny "prawidłowe"
- HbA (HbA1) (2α2β) – prawidłowa hemoglobina dorosłych
- HbA2 (2α2δ) – prawidłowa hemoglobina dorosłych; stanowi około 1,5% – 3% hemoglobiny
- HbF (2α2γ) – hemoglobina płodowa; ma większe powinowactwo do tlenu niż HbA, dzięki czemu jest w stanie pobrać tlen z krwi matki przez
łożysko
i uwolnić go w tkankach płodu. W życiu pozamacicznym jest zastępowana, gdyż słabiej uwalnia
tlen
w tkankach przy wyższym ciśnieniu parcjalnym tlenu. U dorosłych do 2%
- hemoglobiny embrionalne – mają podobne właściwości jak HbF:
- Hemoglobina Gower 1 (ξ2ε2)
- Hemoglobina Gower 2 (α2ε2)
- Hemoglobina Portland (ξ2γ2)
- HbA1c – HbA z przyłączoną nieenzymatycznie, trwale cząsteczką
glukozy
do
N-końcowych
aminokwasów
łańcuchów globiny. Duże stężenie świadczy o proporcjonalnie podwyższonej
glikemii
, co może pozwolić na określenie średniego poziomu glukozy w surowicy przez okres 2-3 miesięcy, a to ma znaczenie np. w ocenie skuteczności leczenia
cukrzycy
. Norma stanowi 4-6% ogólnej ilości hemoglobiny. Produkty przejściowe pomiędzy HbA1 a HbA1c stanowią formy HbA1a oraz HbA1b będące
zasadami Schiffa
, w których glukoza jest przyłączona odwracalnie. Ogólna liczba wszystkich form glikowanej hemoglobiny powinna mieścić się w zakresie 6-8% ogólnej ilości hemoglobiny.
Hemoglobiny nieprawidłowe
- HbS -
mutacja punktowa
– podmiana hydrofilowego kwasu glutaminowego w pozycji A2 (6β) na hydrofobową walinę co powoduje powstanie lepkich miejsc i tworzenia agregatów nieutlenowanej HbS, które zniekształcają erytrocyty prowadząc do
niedokrwistości sierpowatokrwinkowej
- HbM – mutacja powodująca zamianę
histydyny
w pozycji F8 na
tyrozynę
, która stabilizuje żelazo w
hemie
w formie Fe3+ zamiast Fe2+. Hemoglobina z Fe3+ nazywa się methemogolobiną (metHb) i nie wiąże się z tlenem.
- Hemoglobina typu Chesapeake – zamiana
argininy
na
leucynę
w pozycji FG4 (92 w łańcuchu α)
- Hemoglobina typu Bristol – zmiana
waliny
na
kwas asparaginowy
w pozycji 67 łańcucha β. Zmiana nie powoduje zaburzenia funkcji
- Hemoglobina typu Sydney – zmiana waliny na
alaninę
w pozycji 67 łańcucha β. Zmiana nie powoduje zaburzenia funkcji
- Hemoglobina typu Hikari – zmiana
lizyny
na
asparaginę
w pozycji 61 łańcucha β. Zmiana nie powoduje zaburzenia funkcji
- Hemoglobina typu Milwaukee – zmiana waliny na
kwas glutaminowy
w pozycji 67 łańcucha β. Zmiana nie powoduje zaburzenia funkcji
- Hemoglobina typu Lepore – (2α2Lepore) – hemoglobina w jednym z typów
β-talasemii
, wynik
delecji
genów kodujących łańcuchy β i δ. Ich resztki tworzą gen kodujący łańcuch Lepore.
Uwaga: litery greckie w nawiasach oznaczają jakie łańcuchy globiny wchodzą w skład cząsteczki.
Zmiany konformacji pod wpływem tlenu
Hemoglobina. Przechodzenie między stanami T (na animacji stan "deoxy") oraz R (na animacji stan "oxy")
Przyłączenie cząstki tlenu do jednej z czterech cząstek hemu hemoglobiny powoduje zmianę struktury drugo-, trzecio- i czwartorzędowej całego tetrameru. Przyczyną jest wsunięcie atomu
żelaza
(położonego w przypadku nieutlenowanej hemoglobiny w odległości około 0,06 nm od płaszczyzny hemu) w płaszczyznę pierścienia hemu po połączeniu z tlenem.
Wsunięcie atomu żelaza pociąga związaną z nim tzw.
histydynę
proksymalną leżącą w pozycji F8, co powoduje przemieszczenie sąsiednich
aminokwasów
globiny. Doprowadza to w rezultacie do pęknięcia wiązań poprzecznych pomiędzy
końcami karboksylowymi
wszystkich czterech cząstek globiny. W efekcie dochodzi do rotacji pary α1/β1 względem pary α2/β2 o 15°
Konformacja, w której hemoglobina jest tylko częściowo utlenowana, określana jest jako stan T (od
ang.
taut – naprężony), zaś konformacja całkowicie utlenowanej hemoglobiny po wykonaniu obrotu o 15° – jako stan R (ang. relaxed – rozluźniony).
Hemoglobina w stanie R wykazuje większe powinowactwo do tlenu aniżeli w stanie T. W związku z tym każde przyłączenie w płucach cząstki tlenu do hemoglobiny ułatwia przyłączanie następnych (tzw. wiązanie kooperacyjne), zaś odczepienie każdej cząstki tlenu w tkankach ułatwia uwalnianie kolejnych cząstek O2. Wiązanie kooperacyjne sprzyja maksymalizacji wysycania tlenem hemoglobiny w płucach (przy danym ciśnieniu parcjalnym tlenu – PO2) oraz oddawania przez nią tlenu w tkankach.
Wiązanie dwutlenku węgla
Hemoglobina transportuje około 15% z ogólnej ilości CO2 przenoszonego przez krew. W momencie gdy z cząstki hemoglobiny zostaje uwolniony tlen,
dwutlenek węgla
wchodzi w reakcję z grupą α-aminową globiny, tworząc
karbaminian
, jednocześnie w wyniku tej reakcji powstają wolne
protony
równoważące protony zużywane w płucach przez
efekt Bohra
(dwa mole protonów na każdy mol CO2). W wyniku powstania karbaminianu zmienia się ładunek grup na końcach łańcuchów globiny, co umożliwia tworzenie wiązań poprzecznych i przejście do stanu T.
W płucach natomiast, w momencie gdy dochodzi do przejścia ze stanu T do stanu R, w wyniku rozerwania wiązań poprzecznych uwolnione zostają protony z atomów
azotu
pierścieni
imidazolowych
histydyny
znajdującej się w pozycji HC3 (His 146) na łańcuchach β. Łączą się one z
wodorowęglanami
, co prowadzi do powstania
kwasu węglowego
, rozkładanego następnie przez
anhydrazę węglanową
zawartą w erytrocytach na wodę i dwutlenek węgla usuwany z krążenia.
W tkankach występuje niższe
pH
niż w płucach, w związku z czym protony wiążą się z histydyną HC3, sprzyjając przejściu hemoglobiny ze stanu R do stanu T.
Rola bisfosfoglicerynianu
W warunkach niedotlenienia w organizmie zwiększa się synteza
2,3-bisfosfoglicerynianu
(BPG). Substancja ta, powstająca z
1,3-bisfosfoglicerynianu
będącego produktem pośrednim
glikolizy
, ma właściwość wiązania się z tetramerem hemoglobiny znajdującym się w stanie T i stabilizowania go.
W stanie T konformacja łańcuchów hemoglobiny umożliwia wniknięcie między nie jednej cząstki BPG, która następnie wytwarza po trzy (w sumie sześć) wiązań poprzecznych z każdym z łańcuchów β, a dokładniej z posiadającymi dodatni ładunek resztami
waliny
w pozycji NA1,
lizyny
EF6 oraz
histydyny
H21. Te dodatkowe wiązania utrudniają przejście ze stanu T o niższym powinowactwie do tlenu do stanu R, w którym hemoglobina jest mniej skłonna do uwalniania tlenu.
Ciekawym przystosowaniem ewolucyjnym jest zamiana histydyny H21 na
serynę
w łańcuchu γ, który zastępuje łańcuch β w hemoglobinie płodowej HbF. Dzięki temu BPG ma mniejszy wpływ na hemoglobinę płodową, a co za tym idzie przy niższym stężeniu tlenu HbF ma do niego większe powinowactwo niż HbA. Efekt ten umożliwia wymianę gazową między krwią płodu a krwią matki zachodzącą w
łożysku
.
Normy
Normy ilości hemoglobiny we krwi dorosłego człowieka wynoszą około 11,0-17,5 g/dl, jednak ze względu na różne metody pomiarowe każde laboratorium analityczne ustala własne normy (zwykle podyktowane przez producenta analizatora). Należy również zaznaczyć, że fizjologicznie stężenie hemoglobiny u mężczyzn jest wyższe niż kobiet.
Pochodne hemoglobiny
Zobacz też